

Mole, J;
Mead, S;
Rudge, P;
Nihat, A;
Mok, T;
Collinge, J;
Caine, D;
(2021)
Cognitive decline heralds onset of symptomatic inherited prion disease.
Brain
, 144
(3)
pp. 989-998.
10.1093/brain/awaa409.

Preview |
Text (Article)
Mole_converters neuropsychology paper revision_clean.pdf - Accepted Version Download (255kB) | Preview |
![[thumbnail of Figure 1]](https://discovery.ucl.ac.uk/10129069/7/Mole_Figure%201.png)  Preview |
Image (Figure 1)
Mole_Figure 1.png Download (261kB) | Preview |
Preview |
Image (Figure 2)
Mole_Figure 2.tif Download (1MB) | Preview |
Preview |
Image (Figure 3)
Mole_Figure 3.tif Download (1MB) | Preview |
Preview |
Image (Figure 4)
Mole_Figure 4.tif Download (1MB) | Preview |
Preview |
Image (Figure 5)
Mole_Figure 5.tif Download (1MB) | Preview |
Preview |
Text (Figure legends)
Mole_Figure legends_clean.pdf Download (8kB) | Preview |
Preview |
Text (Supplementary material)
Mole_Supplementary Material_clean.pdf Download (169kB) | Preview |
Abstract
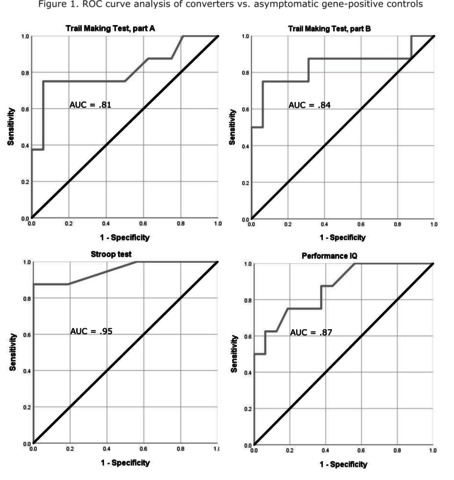
The clinical effectiveness of any disease-modifying treatment for prion disease, as for other neurodegenerative disorders, will depend on early treatment before damage to neural tissue is irrevocable. Thus, there is a need to identify markers that predict disease onset in healthy at-risk individuals. Whilst imaging and neurophysiological biomarkers have shown limited use in this regard, we recently reported progressive neurophysiological changes in individuals with the inherited prion disease mutation P102L. We have also previously demonstrated a signature pattern of fronto-parietal dysfunction in mild prion disease. Here we address whether these cognitive features anticipate the onset of symptoms in a unique sample of patients with inherited prion disease. In the cross-sectional analysis, we analysed the performance of patients at three time points in the course of disease onset: prior to symptoms (n = 27), onset of subjective symptoms without positive clinical findings (n = 8) and symptomatic with positive clinical findings (n = 24). In the longitudinal analysis, we analysed data from 24 patients who were presymptomatic at the time of recruitment and were followed up over a period of up to 17 years, of whom 16 remained healthy and eight converted to become symptomatic. In the cross-sectional analysis, the key finding was that, relative to a group of 25 healthy non-gene carrier controls, patients with subjective symptoms but without positive clinical findings were impaired on a smaller but similar set of tests (Trail Making Test part A, Stroop test, Performance IQ, gesture repetition, figure recall) to those previously found to be impaired in mild prion disease. In the longitudinal analysis, Trail Making Test parts A and B, Stroop test and Performance IQ scores significantly discriminated between patients who remained presymptomatic and those who converted, even before the converters reached criteria for formal diagnosis. Notably, performance on the Stroop test significantly discriminated between presymptomatic patients and converters before the onset of clinical symptoms [area under the curve = 0.83 (95% confidence interval, 0.62–1.00), P = 0.009]. Thus, we report here, for the first time, neuropsychological abnormalities in healthy patients prior to either symptom onset or clinical diagnosis of inherited prion disease. This constitutes an important component of an evolving profile of clinical and biomarker abnormalities in this crucial group for preventive medicine.

{kind=link}
{kind=link}
Archive Staff Only
 |
View Item |